(Registered under the Ministry of SME, Government of India for Research and Experimental Development on Natural Sciences and Engineering) |UDYAM-DL-06-0096855|
ISO 9001: 2015 Certification: Accredited by United Ackreditering Services Limited, United Kingdom |QMS/C26E/0923|
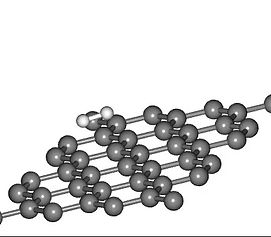
Hydrogen Molecule Adsorption study on Graphene sheet using DFT Model

Gold Nanocluster Density Functional Theory Structural Optimization
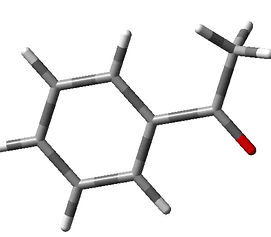
DFT/B3LYP/6-311G*(d,p) Computed Vibrational Modes in Acetophenone
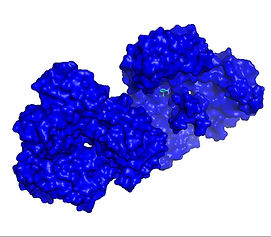
Molecular Docking of CD30 Peptide (TNF Receptor Associated Factor 2 Aspirin

Molecular Dynamics Simulation: NPT Equlibration of CD30 Protein
Remarkable Achievements the Way




































Advance Your Scientific Skills with Our Skillfully Designed Training Programs.




The Statistics demonstrate our Achievements.
We Appreciate you being part of our Journey!




-
Dr. Susantha Ganegamage, Assistant Professor at Lamar University, Texas, United States of America (USA) DFT-G
-
Dr. Binod R Giri, Senior Research Scientist at King Abdullah University of Science and Technology (KAUST), Saudi Arabia DFT-G
-
Dr. Shaza Massarani, Professor (Full) at King Saud University (KSU), Saudi Arabia DFT-G
-
Dr. Aziz Unnisa, Associate Professor at the University of Hail (UOH), Saudi Arabia DFT-G
-
Dr. Abdel-Baset H. Mekky, Professor (Associate), Qassim University (QU), Saudi Arabia DFT-G
-
Dr. Hela Ferjani, Professor (Associate), Imam Mohammad ibn Saud Islamic University (IMSU), Saudi Arabia DFT-G
-
Dr. Felipe Cordova LozanoFundación, Professor, Universidad de las Américas-Puebla (UAP), Mexico DFT-G
-
Dr. Murali Venkata Basavanga Unnamatla, Fulltime Professor, Universidad Autónoma del Estado de México - Inicio (UEE), Mexico DFT-G
-
Dr. Jose Manuel Bravo-Arredondo, Professor (Associate), Autonomous University of Tlaxcala (UOT), Mexico DFT-G
-
Dr. Syed Shaheen Shah, Assistant Professor, Kyoto University (KU), Japan DFT-G
-
Dr.Elhassane Mohamed Abdssalam Anouar, Associate Professor, Prince Sattam bin Abdulaziz University (PSAU), Saudi Arabia DFT-G
-
Prof. (Dr.) Amartya Sengupta, Professor, Indian Institute of Technology (IIT) Delhi, Delhi, India DFT-G
-
Prof. (Dr.) Arindam Sarkar, Professor, Indian Institute of Technology (IIT) Bombay, Maharashtra, India RRD
-
Prof. (Dr.) Chandramohan Palogi, Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India RRD
-
Prof. (Dr.) Manikandan P, Professor, Indira Gandhi Centre for Atomic Reserach (IGCAR), Kalpakkam, Tamil Nadu, India RRD
-
Prof. (Dr.) Rajesh Prasad, Professor, Indian Institute of Technology (IIT) Delhi, Delhi, India RRD
-
Prof. (Dr.) Santu Dey, Professor, Variable Energy Cyclotron Centre, India DFT-M
-
Prof. (Dr.) Subrato Bhattacharya, Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-G
-
Prof. (Dr.) Vinod Kumar Kannaujiya, Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-G
-
Prof. (Dr.) Krishna Kumar Singh, Professor, Birla Institute of Technology and Science (BITS) Pilani, Dubai, United Arab Emirates DFT-M
-
Prof. (Dr.) Prashant Kharkar, Professor, Institute of Chemical Technology (ICT), Mumbai, Maharashtra, India CADD
-
Prof. (Dr.) Bivas Dam, Professor, Jadavpur University (JU), West Bengal, India RRD
-
Prof. (Dr.) Ozair Alam, Professor, Jamia Hamdard University (JHU), New Delhi, India CADD
-
Prof. (Dr.) Satyajit Banerjee, Professor, Indian Institute of Technology (IIT) Kanpur, India DFT-M
-
Prof. (Dr.) Parnika Das, Professor, Homi Bhabha National Institute (HBNI), India DFT-M
-
Dr. Sandeep Pokharia, Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-M
-
Dr. Surendra Singh, Associate Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India DFT-M
-
Dr. Rajeev Kumar, Associate Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India DFT-M
-
Dr. R C Das, Associate Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India DFT-M, DFT-M_A, RRD
-
Dr. Ajish Kumar K S, Assistant Professor, Bhabha Atomic Research Centre (BARC), Maharashtra, India DFT-M
-
Dr. Abhijeet L. Sangle, Assistant Professor, Indian Institute of Technology (IIT) Bombay, Maharashtra, India RRD, DFT-M
-
Dr. Apurav Guleria, Assistant Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India DFT-M, RRD
-
Dr. Bani Mahanti, Assistant Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-G
-
Dr. D N V V Konda Lutukurthi, Assistant Professor, Indian Institute of Technology (IIT-ISM) Dhanbad, Jharkhand, India DFT-G, DFT-M
-
Dr. Debasish Manna, Assistant Professor, Indian Institutes of Science Education and Research (IISER) Bhopal, Madhya Pradesh, India DFT-G
-
Dr. Divya Kushwaha, Assistant Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-G
-
Dr. Kodanda Ram Mangipudi, Assistant Professor, Indian Institute of Technology (IIT) Bhubaneswar, Odisha, India RRD
-
Dr. R Lalneihpuii, Assistant Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-G
-
Dr. Rahul Sharma, Assistant Professor, Indian Institutes of Science Education and Research (IISER) Berhampur, Odisha, India DFT-M
-
Dr. Sandeep Patel, Assistant Professor, Banaras Hindu University (BHU), Uttar Pradesh, India DFT-G
-
Dr. Santanu Mandal, Assistant Professor, Indian Institute of Technology (IIT) Bhubaneswar, Odisha, India DFT-M, DFT-M_A
-
Dr. Selvakumar Jayaprakasam, Assistant Professor, Homi Bhabha National Institute (HBNI), Tamil Nadu, India DFT-M
-
Dr. Sonal Shrivastava, Assistant Professor, Indian Institute of Technology (IIT) Patna, Bihar, India RRD
-
Dr. Sree Rama Murthy Anupindi, Assistant Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India DFT-G
-
Dr. Subhas Samanta, Assistant Professor, Indian Institute of Technology (IIT) Jammu, Jammu and Kashmir DFT-G
-
Dr. Sumit Kamal, Assistant Professor, Indian Institute of Technology (IIT) Jodhpur, Rajasthan, India DFT-G, DFT-M
-
Dr. Supriyo Ghosh, Assistant Professor, Indian Institute of Technology (IIT) Roorkee, Uttarakhand, India DFT-M
-
Dr. Sushil Kumar, Assistant Professor, Homi Bhabha National Institute (HBNI), Maharashtra, India DFT-G
-
Dr. Veeramani Chidambaranathan, Assistant Professor, Indian Institute of Technology (IIT) Roorkee, Uttarakhand, India DFT-M
-
Dr. Venkatadivakar Botcha, Assistant Professor, BioSense Institute, University of Novi Sad, Novi Sad, Serbia DFT-
Thank you to the Top 50 Faculty for attending our Training! Your commitment is Inspiring
Unleashing Potential: Experts' Perspectives
Centre hosted four Keynote Lectures by the renowned Computational Chemistry Researchers from the Prestigious Research Institutions of the world. These sessions offered valuable insights and encouraged dynamic discussions on the latest developments in the field. We are dedicated to promoting knowledge exchange and collaboration within the scientific community.




Driving Change Through Impactful Ideas




Dr. Nikhil Aggarwal is a well-regarded academician known for delivering a multitude of online lectures as an Invited Speaker at various Research Institutions. His dynamic presentation style and deep knowledge have a considerable influence on students and professionals, offering valuable insights in his field of expertise.























